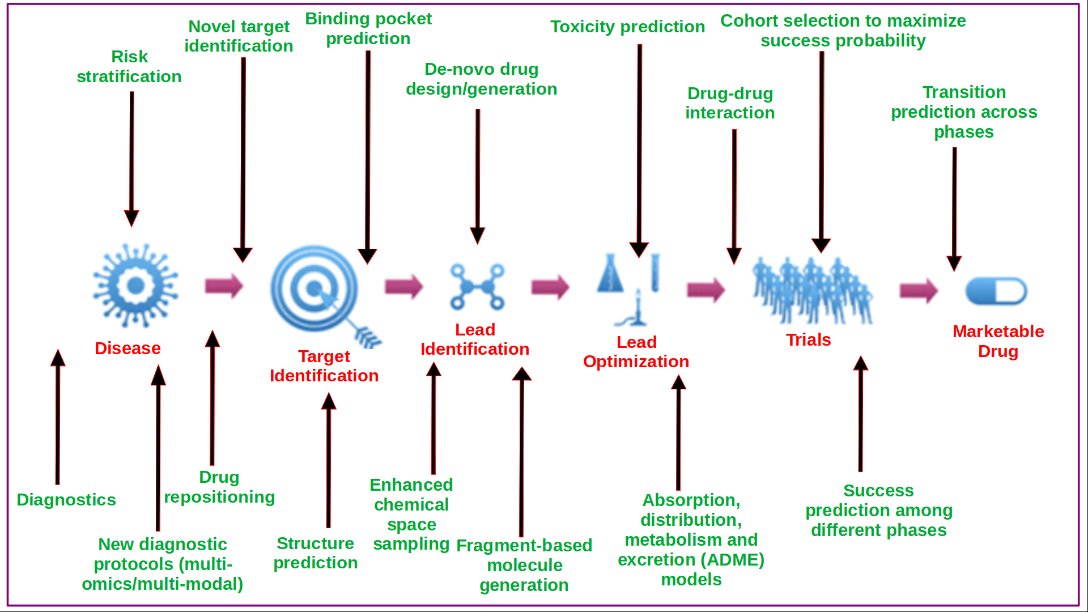
Data Driven Drug Discovery (D4)
Drug Discovery Process
Datasets
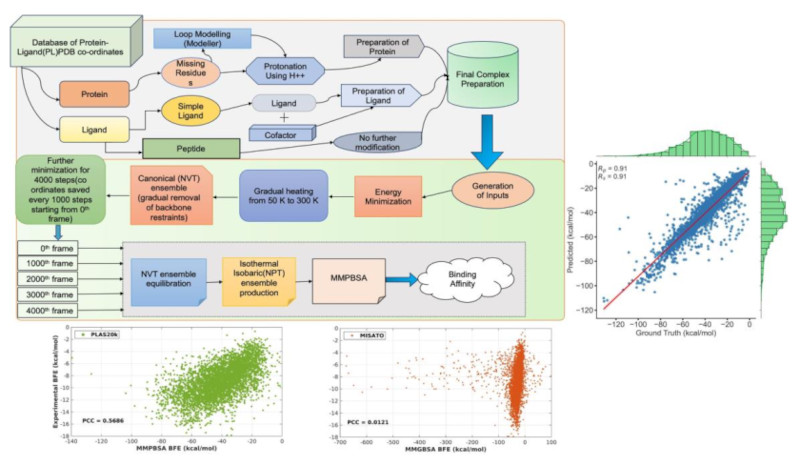
The rapid advancement of computing technologies, particularly AI, has transformed numerous fields, including drug discovery. ML is pivotal throughout drug development, but accessing high- quality data remains challenging due to cost constraints. To address this, researchers are turning to computational methods for data generation. Dynamic structures are crucial for guiding compound design and evaluating protein-ligand interactions, yet existing datasets lack such data. IIIT-H/IHub-Data aims to create the largest collection (~20000 protein ligand complexes (PLC)) of molecular dynamics trajectories. Initially this effort was started by creating a dataset of 5k PLCs (PLAS-5K). This was further extended to ~20 PLCs (PLAS-20K). This dataset is therefore enriched with information of binding energy scores corresponding to each dynamic coordinates generated for each PLC. A correlation of 0.53 is obtained with the corresponding ground truth binding affinity for the experimental PLCs (~ 6600) within the dataset
Variation 01
This version of the datasets have the refined crystal structures of protein-ligand complexes, in explicit solvent environment, ready for simulations. Structures sourced from the Protein Data Bank (PDB) are carefully processed to retain only the biologically relevant chains involved in complex formation. Additionally, any missing loop regions within these structures are filled to ensure completeness and accuracy. We have provided the binding free energy from MMPBSA using trajectories from unbiased MD simulations along with the energy components. These datasets provide detailed understanding of the energy components involved in binding that can aid in the development and validation of computational models for predicting binding affinities, as well as in unraveling the molecular mechanisms underlying ligand binding
Variation 02
In this version of the datasets, dynamic information has been incorporated, enriching them with detailed insights into protein-ligand interactions. Specifically, each entry in the dataset comprises information on protein-ligand complexes alongside two explicitly chosen water molecules. These water molecules are selected dynamically using an in-house code, which identifies the two closest water molecules to both the protein and ligand. The dataset includes simulated trajectories, capturing the dynamic behavior of the protein-ligand-water system over time. Notably, each PDB ID entry contains five independent trajectories. Additionally, the dataset features binding free energy calculations for each frame of the trajectories, computed using the Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA) method. By integrating dynamic information and binding free energy calculations, the datasets offer a comprehensive resource for researchers interested in studying protein-ligand interactions.
Variation 3
These versions of the datasets offer a diverse collection of protein-ligand complexes featuring solvated environment and stand out as superior to its predecessors, variations 1 and 2, due to its provision of complete trajectories. What sets this dataset apart is its uniqueness in providing binding affinities for each frame, alongside average values, enriching the understanding of molecular interactions. Each entry in the dataset corresponds to a PDB ID and includes 200 frames, with 40 frames sourced from each of the five independent runs. The inclusion of complete trajectories and binding affinity data is invaluable for researchers in computational biology and drug discovery, offering a rich resource for studying protein-ligand interactions in solvated environments
PLAS 5K
- Variation 01 : https://datafoundation.iiit.ac.in/dataset-versions/453982e4-3ff8-4785-8d49-f887c8f4b48a
- Variation 02 : https://datafoundation.iiit.ac.in/dataset-versions/547d94d3-0a34-4c3c-9f2e-a5cfa8ecad4d
- Variation 03 : https://datafoundation.iiit.ac.in/dataset-versions/712d3a07-8164-4a87-bc58-7c0d7fbdfb6d
- PLAS 20K (Versions 01 & 02) : https://datafoundation.iiit.ac.in/dataset-versions/6f6c118b-d4f3-4877-9738-500674094e45
Machine Learning Models
Improving Protein-Ligand Binding Affinity Prediction using Deep Learning and Molecular Dynamics Trajectories
Traditional drug discovery is hindered by high costs, time constraints, and low success rates, highlighting the need for improved initial binding affinity prediction methods. Machine Learning scoring functions show promise in predicting binding affinity but struggle with biases in virtual screenings. Additionally, most datasets lack dynamic information, limiting accuracy.
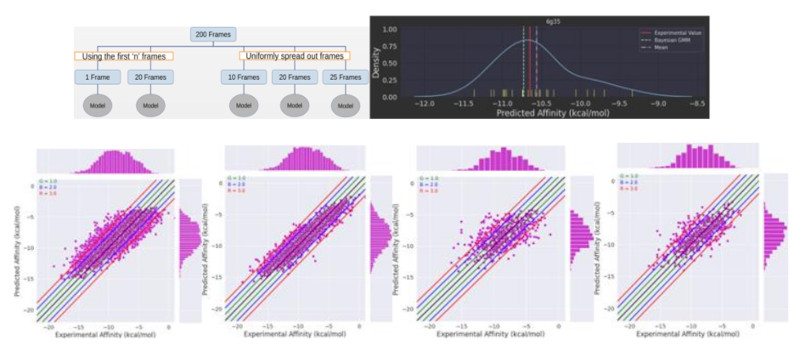
Graph-based deep learning, particularly using molecular dynamics (MD) studies, offers more accurate predictions. Therefore, we propose utilizing synthetic datasets like PLAS- 20k, containing 20,000 protein-ligand complexes (PLCs) with MD simulations, to enhance prediction accuracy and generalizability. Our study employs the deep neural network, Pafnucy, to predict the binding affinity of ~6180 PLCs from the PLAS-20k dataset, observing enhanced results with dynamic information incorporation. We show that training the model with varied dynamic data improves generalizability, underscoring the importance of dynamic dataset augmentation.
BandNN
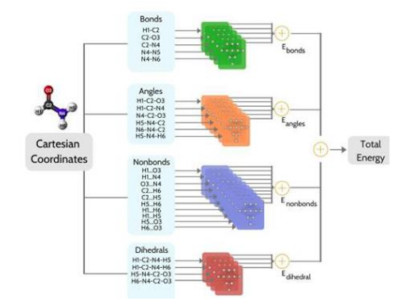
BandNN presents a transferable and molecule size-independent neural network (NN) that models bonds (B), angles (A), non-bonded (N) interactions, and dihedrals (D) (BANDNN) based on chemically intuitive representation inspired by molecular mechanics force fields. The model predicts the atomization energies of equilibrium and non-equilibrium structures as sum of energy contributions from various components.
Link to github: https://github.com/devalab/BAND-NN
CIGIN
ChemicallyInterpretableGraphInteractionNetwork (CIGIN) is a machine learning model driven by graph neural networks that predicts the solvation free energy of drug like molecules or solutes. CIGIN trained on the FREESOLV dataset yields an RMSE of 0.91 kcal/mol which is comparable to the state-of-the-art method while exhibiting better transferability to unknown solute-solvent combinations.
Link to github : https://github.com/devalab/CIGIN
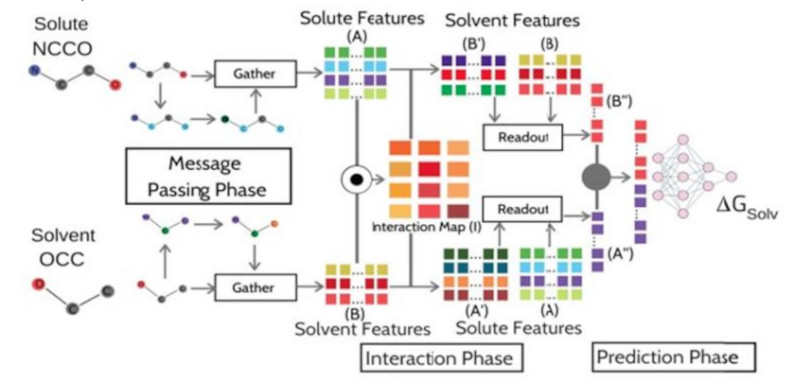
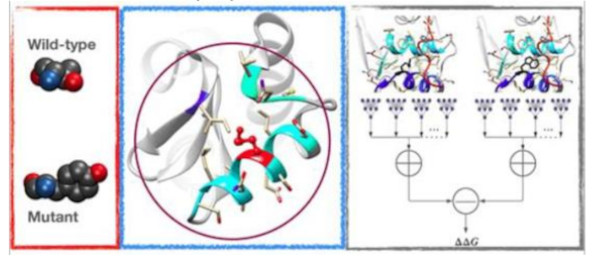
SCONES
Self-COnsistentsNEural network proteinStability prediction (SCONES) is an interpretable neural network that estimates a residue's contribution towards protein stability(ΔG)in its local structural environment. It is a first of its kind machine learning model that proposes transitive data augmentation, evaluating transitive consistency that incorporates both symmetric and transitive properties into its architecture.
Link to github : https://github.com/devalab/SCONES
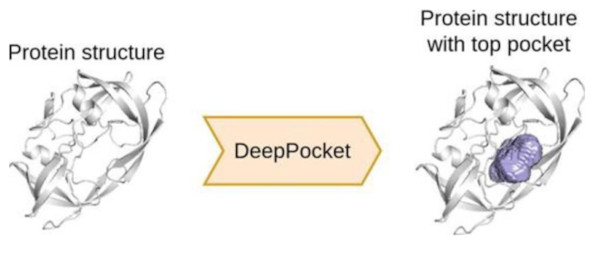
Deeppocket
A structure-based drug design pipeline involves the development of potential drug molecules or ligands that from stable complexes with a given receptor at its binding site. As a combination of geometry-based software and deep learning, we report a novel framework, DeepPocket that utilizes 3D convolutional neural networks for the rescoring of pockets identified by Fpocket and further segments these identified cavities on the protein surface.
Link to github : https://github.com/devalab/DeepPocket
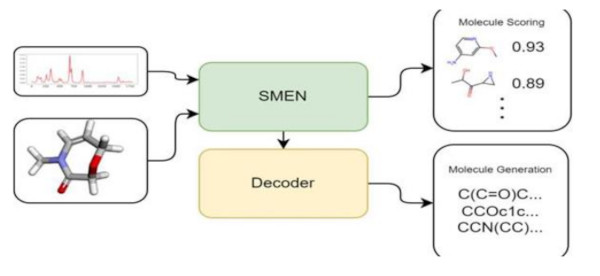
Spectra to Structure: Contrastive Learning Framework for Library Ranking and Generating Molecular Structures
Inferring complete molecular structure from Infrared (IR) spectra is a challenging task. In this work, we propose SMEN (Spectra and Molecule Encoder Network), a framework for scoring molecules against given IR spectra. The proposed framework uses contrastive optimization to obtain similar embedding for a molecule and its spectra. Using the proposed method, we can rank the molecules in the ǪM9 Dataset using embedding similarity and obtain a Top 1 Accuracy of∼81 percent, Top 3 accuracy of∼96 percent, and Top 10 accuracy of ∼99 percent on the evaluation set. We extend SMEN to build a generative transformer for a direct molecule prediction from IR spectra. The proposed method can significantly help molecule library ranking tasks and aid the problem of inferring molecular structures from spectra
Link to Github: https://github.com/devalab/Spectra2Structure
MoleGuLAR
While methods like high throughput screening and virtual screening require extensive searches in large databases, MoleGuLAR proposes a conditional drug generation framework using Stack Augmented RNNs optimized using multi- objective rewards. The framework is able to generate drug like molecules with high binding affinities to specific targets.
Link to Github: https://github.com/devalab/MoleGuLAR

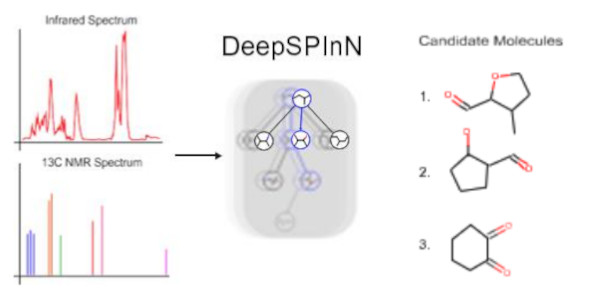
DeepSPInN
DeepSPInN is a deep reinforcement learning method that predicts the molecular structure when given Infrared and 13C Nuclear magnetic resonance spectra by formulating the molecular structure prediction problem as a Markov decision process (MDP) and employs Monte Carlo tree search to explore and choose the actions in the formulated MDP. On the ǪM9 dataset, DeepSPInN is able to predict the correct molecular structure for 91.5% of the input spectra in an average time of 77 seconds for molecules with less than 10 heavy atoms.
Link to Github : https://github.com/devalab/DeepSPInN
Amyloid Hotspot Prediction using Machine Learning
Amyloidosis is a disorder that causes a buildup of amyloid fibrils in the body. This is a disorder associated with many life- threatening diseases like Alzheimer's, Diabetes, etc. thus emphasizing the necessity to detect these sequences within the proteome. Our model uses known protein sequences that are hot-spot regions for aggregation using Attention mechanism that powers transformers along with protein physicochemical properties to predict such sequences. Our Model currently achieves 0.91 AUC with 200x lower Inference times compared to previous Deep Learning model.

Graph Neural Networks for Accurate Prediction of Drug- Drug Interactions
Drug-Drug Interactions (DDI) prediction is critical in drug discovery due to the risks of adverse drug events and the increase in polypharmacy. This work introduces an ML approach using Graph Neural Networks (GNN) to accurately predict DDIs. The method involves three stages: (1) Featurization, where GNN extracts atomic features of drugs, (2) Interaction, calculating interaction maps between drug atom pairs, and (3) Readout, combining these maps and features for a unified drug representation, followed by a feed-forward neural network for DDI prediction. The model excels in predicting the presence and types of DDIs, outperforming existing models with an F1 score of 0.98 for DDI prediction and 0.90 for categorizing DDI event types.
Link to Github : https://github.com/devalab/GraphDDI
